Serghei Mangul and Lana Martin, together with Eleazar Eskin, recently wrote a paper describing a model for training undergraduates in Bioinformatics. Our paper is available online as a preprint and is under review at a peer-reviewed journal.
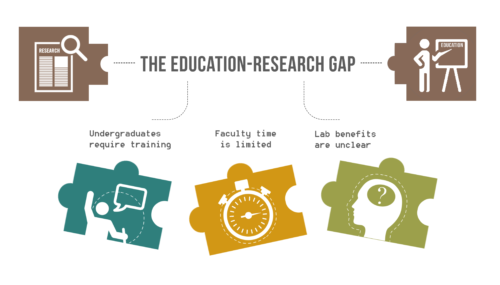
The Education-Research Gap in Universities.
While the benefits of undergraduate research experiences (UREs) are recognized for undergraduates, the advantages of UREs for graduate students, post-doctoral scholars, and faculty are not clearly outlined.
Based on our experience mentoring undergraduates in ZarLab, we believe that the analysis of genomic data is particularly well-suited for successful involvement of undergraduates. In computational genomics research, undergraduate trainees who master a particular skill can contribute sufficient work to gain authorship on a peer-reviewed paper.
In our paper, we offer a framework for engaging undergraduates in genomics research while simultaneously improving lab productivity: first, identify particular “low-level” tasks that may take up to a week for an undergraduate to complete. Second, encourage students to “outsource” foundational education needs with workshops, online resources, and review articles. Third, genomics research labs can take advantage of department- and campus-wide undergraduate research and training initiatives.
The proposed strategy can be easily reproduced at other institutions, is pedagogically flexible, and is scalable from smaller to larger laboratory sizes. We hope that genomics researchers will involve undergraduates in more computational tasks that benefit both students and senior laboratory members.
Preprint copies of our manuscript are available for download here: https://peerj.com/preprints/3149/
In tandem with this paper, we created an online catalogue of resources and papers aimed at bridging the research-teaching divide in computational genomics: https://smangul1.github.io/undergraduates-in-genomics/
The full citation of our paper:
Mangul, S., Martin, L. and Eskin, E., 2017. Involving undergraduates in genomics research to narrow the education-research gap. PeerJ Preprints, 5, p.e3149v1.
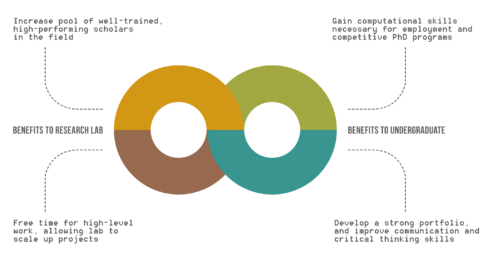
Benefits of UREs to Research Lab and Undergraduates.