Jong Wha (Joanne) Joo developed an approach to simultaneously analyze multiple phenotypes in a genome-wide association studies (GWAS) dataset. She introduces this new methodology, referred to as GAMMA (Generalized Analysis of Molecular variance for Mixed model Analysis), in a paper recently published in Genetics.
GWASs have identified many genetic variants involved in traits and development of human diseases by examining for correlation of a single phenotype and individual genotype one phenotype at a time. Since initial development of the standard GWAS approach, GWAS data collection has become larger in scale and higher in resolution. Today’s large-scale datasets include expression data and often contain thousands of phenotypes per individual. Performing the standard single-phenotype analysis on these datasets is slow and potentially fails to detect unmeasured aspects of complex biological networks.
Analyzing many phenotypes simultaneously increases the power to detect more variants and capture previously unmeasured aspects of the genome. However, standard GWAS approaches capable of simultaneously testing multiple phenotypes fail to account for the distorting effects of population structure, a phenomenon present in large cohorts that inevitably contain individuals sharing common ancestry from multiple populations. As a result, standard GWAS approaches either fail to detect true effects or produce many false positive identifications.
GAMMA is an efficient, robust approach capable of simultaneously analyzing many phenotypes while correcting for population structure. GAMMA uses the principles behind existing linear mixed models to analyze for many phenotypes simultaneously and a multiple regression technique to correct for population structure.
Joanne’s paper presents the results of testing GAMMA for accuracy in three scenarios: a simulated dataset containing population structure, a yeast dataset containing many trans-regulatory hotspots, and a complex gut microbiome dataset. In the simulated study using data implanted with true population structure effects, GAMMA accurately identifies these true effects without producing false positives. In the simulation with yeast data, GAMMA successfully corrected for the bias of technical artifacts such as batch effects and identified significant signals on most of the putative hotspots. In the third test, Joanne and her team assesses GAMMA’s ability to perform a multiple-phenotypes analysis with microbiome data. Here, results identified nine loci likely to have true biological mechanisms in the taxa.
In each scenario, results of GAMMA were compared to those of the standard t-test, EMMA, and MDMR. The standard t-test and EMMA failed to identify true variants, because the phenotypic effects in each example is smaller than the amount these methods are powered to detect. MDMR produced no significant signals in the yeast dataset and identified many false associations in the simulated and gut microbiome datasets. Both GAMMA and MDMR have sufficient power to detect small association signals in these complex datasets, but only GAMMA successfully corrects for population structure.
This project was led by Joanne Joo and involved Eun Yong Kang and Farhad Hormozdiari. The article is available at: http://www.genetics.org/content/204/4/1379.
GAMMA was developed by Joanne Joo, Eun Yong Kang, Elin Org, Nick Furlotte, Brian Parks, Aldons J. Lusis, and Eleazar Eskin. Visit the following page to download GAMMA: http://genetics.cs.ucla.edu/GAMMA/
The full citation to our paper is:
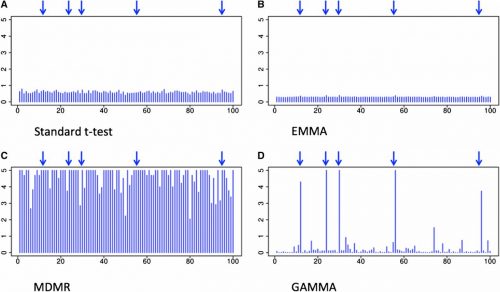
The results of GAMMA and three standard GWAS methods applied to a simulated dataset. The x-axis shows SNP locations and the y-axis shows log10p-value of associations between each SNP and all the genes. Blue arrows show the location of the true trans-regulatory hotspots.