
Computational Genomics Summer Institute
CGSI is a unique opportunity for scholars to foster relationships and unleash the full potential of their projects.

UCLA Computational Genetics



The use of the multivariate normal (MVN) model has been a powerful tool in our groups research and it has been utilized in many of our papers. Jose Lozano (University of the Basque Country, San Sebastian, Spain), along with Eleazar Eskin and three ZarLab alumni—Farhad Hormozdiari (postdoc at Harvard), Jong Wha (Joanne) Joo (faculty at Dongguk University in Seoul), and Buhm Han (faculty at University of Ulsan College of Medicine in Seoul)—recently published a review of the multivariate normal (MVN) distribution framework in genome-wide association studies (GWAS) studies.
Genome-wide association studies (GWAS) have discovered thousands of variants involved in common human diseases. In these studies, frequencies of genetic variants are compared between a population of individuals with a disease (cases) and a population of healthy individual controls). Any variant that has a significantly different frequency between the two populations is considered an associated variant.
A major challenge in the analysis of GWAS studies is the fact that human population history causes nearby genetic variants in the genome to be correlated with each other. In this review, we demonstrate how to utilize the MVN distribution to explicitly take into account the correlation between genetic variants and provide a comprehensive framework for analysis of GWAS.
In this paper, we show how the MVN framework can be applied to perform association testing, correct for multiple hypothesis, testing, estimate statistical power, and perform fine mapping and imputation. In future blog posts, we will highlight different ways the MVN framework can be used in association studies.
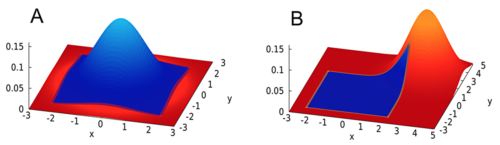
An illustration of the multivariate normal model (a) Type I Error (b) Power.
Many of the authors are the alumni of the group who pioneered the use of the MVN in various problems in association studies. Here is a list of papers that our group published using the MVN framework:
The paper was written by Jose A. Lozano, Farhad Hormozdiari, Jong Wha (Joanne) Joo, Buhm Han, and Eleazar Eskin, and it is available at: https://www.biorxiv.org/content/early/2017/10/28/208199.
The full citation to our paper is:
Jose A. Lozano, Farhad Hormozdiari, Jong Wha (Joanne) Joo, Buhm Han, Eleazar Eskin. 2017. The Multivariate Normal Distribution Framework for Analyzing Association Studies. bioRxiv doi: https://doi.org/10.1101/208199.

On Friday, April 28, 2017, in the CNSI Auditorium, Eleazar Eskin presented ZarLab’s research on fine mapping causal variants and allelic heterogeneity at the 2nd Annual Institute for Quantitative and Computational Biosciences (QCBio) Symposium.
Geneticists use a technique called Genome Wide Association Studies (GWAS) to identify genetic variants that cause an individual to exhibit a particular trait or disease. Typically, GWAS identifies an association signal which suggests that genetic variants within a region of the genome — known as a locus — are associated with the condition. The process of identifying the actual variant in the region which has an affect on the disease is referred to as “fine mapping.”
In addition to finding the actual variants affecting a disease, fine mapping also seeks to address questions that are related to the genetic basis of disease. First, how many causal variants does a locus contain? A disease could be caused by one, single variant or multiple variants that independently affect disease status. We refer to the latter phenomenon as allelic heterogeneity (AH).
Second, when analyzing results from multiple GWASes, can the same causal variant identified in one study be assumed causal in other studies? A GWAS can identify many variants that are associated with two or more traits; however, this correlation can be induced by a confounding factor known as linkage disequilibrium. Colocalization methods seek to identify shared and distinct causal variants.
Farhad Hormozdiari, a recent alumnus of our group and a post-doc at Harvard University, developed several novel approaches for improving the accuracy and efficiency of fine mapping despite presence of AH in the study population. Hormozdiari’s software, CAVIAR, CAVIAR-Genes, and eCAVIAR, are capable of quantifying the probability of a variant to be causal in GWAS and eQTL studies, while allowing for an arbitrary number of causal variants.
In a video of his presentation, Eskin summarizes the progress on these problems. A video of Eskin’s presentation may be found on the QCBio website: https://qcb.ucla.edu/events-seminars/symposium/#toggle-id-2
More details about our research in fine mapping are available in the following papers:
Hormozdiari F, Zhu A, Kichaev G, Ju CJ, Segrè AV, Joo JW, Won H, Sankararaman S, Pasaniuc B, Shifman S, Eskin E. Widespread allelic heterogeneity in complex traits. The American Journal of Human Genetics. 2017 May 4;100(5):789-802.

Serghei Mangul and Lana Martin, together with Eleazar Eskin, recently wrote a paper describing a model for training undergraduates in Bioinformatics. Our paper is available online as a preprint and is under review at a peer-reviewed journal.
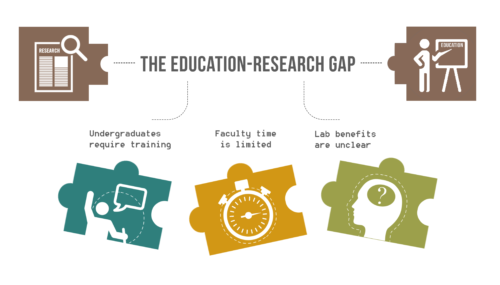
The Education-Research Gap in Universities.
While the benefits of undergraduate research experiences (UREs) are recognized for undergraduates, the advantages of UREs for graduate students, post-doctoral scholars, and faculty are not clearly outlined.
Based on our experience mentoring undergraduates in ZarLab, we believe that the analysis of genomic data is particularly well-suited for successful involvement of undergraduates. In computational genomics research, undergraduate trainees who master a particular skill can contribute sufficient work to gain authorship on a peer-reviewed paper.
In our paper, we offer a framework for engaging undergraduates in genomics research while simultaneously improving lab productivity: first, identify particular “low-level” tasks that may take up to a week for an undergraduate to complete. Second, encourage students to “outsource” foundational education needs with workshops, online resources, and review articles. Third, genomics research labs can take advantage of department- and campus-wide undergraduate research and training initiatives.
The proposed strategy can be easily reproduced at other institutions, is pedagogically flexible, and is scalable from smaller to larger laboratory sizes. We hope that genomics researchers will involve undergraduates in more computational tasks that benefit both students and senior laboratory members.
Preprint copies of our manuscript are available for download here: https://peerj.com/preprints/3149/
In tandem with this paper, we created an online catalogue of resources and papers aimed at bridging the research-teaching divide in computational genomics: https://smangul1.github.io/undergraduates-in-genomics/
The full citation of our paper:
Mangul, S., Martin, L. and Eskin, E., 2017. Involving undergraduates in genomics research to narrow the education-research gap. PeerJ Preprints, 5, p.e3149v1.
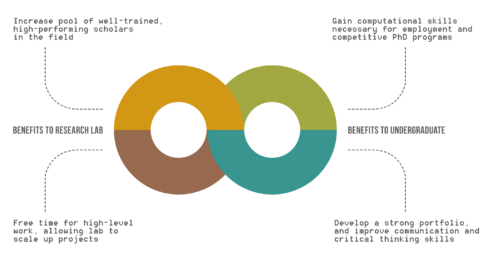
Benefits of UREs to Research Lab and Undergraduates.


