
Our group recently developed the Read Origin Protocol (ROP) method to discover the source of all reads in an RNA-seq experiment. Reads originate from complex RNA molecules, recombinant antibodies and microbial communities. ROP accounts for 98.8% of all reads across poly(A) and ribo-depletion protocols, compared to 83.8% by conventional reference-based protocols. We find that the vast majority of unmapped reads are human in origin and originate from diverse sources, including repetitive elements, non-co-linear elements or recombined B and T cell receptors (BCR/TCR). In addition to human RNA, a large number of reads were microbial in origin, often occurring in sufficient numbers to study the taxonomic composition of microbial communities.
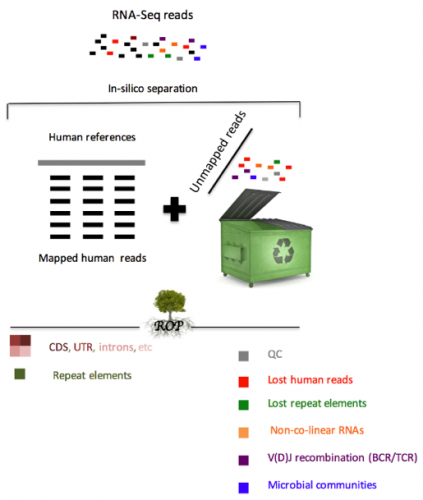
The majority of RNA-Seq analyses begin by mapping each experimentally produced sequence (i.e., read) to a set of annotated reference sequences for the organism of interest. For both biological and technical reasons, a significant fraction of reads remains unmapped. Our study is the first that systematically accounts for almost all reads in RNA-seq studies. We demonstrate the value of analyzing unmapped reads present in the RNA-seq data to better understand the functional mechanisms underlying the connection between immune system, microbiome, human gene expression, and disease etiology.
We applied our method to to RNA-seq data from 53 asthmatic cases and 33 controls collected from three tissues, using both poly(A) selection and ribo-depletion libraries. Using the ROP pipeline we show that immune profiles of asthmatic individuals are significantly different from the controls with decreased T-cell/B-cell receptor diversity and that immune diversity is inversely correlated with microbial load. This case study highlights the potential for novel discoveries without additional TCR/BCR or microbiome sequencing when the information in RNA-seq data is fully leveraged by incorporating the analysis of unmapped reads.
The ROP can not only help researchers make the best use of sequencing data, but will also enable additional scientific questions to be answered with no additional cost. For example, one can now interrogate additional features of the immune system without additional expensive TCR/BCR sequencing. The ‘dumpster diving’ profile of unmapped reads output by our method is not limited to RNA-Seq technology and may be applied to whole-exome and whole-genome sequencing. We anticipate that ‘dumpster diving’ profiling will find broad future applications in studies involving different tissue and disease types.
This project was led by Serghei Mangul and involved Harry Yang (Taegyun), both of whom developed the protocol as open source software. This was a joint project with the Noah Zaitlen group (http://zaitlenlab.ucsf.edu/) at University of California, San Francisco.
ROP is available at https://sergheimangul.
The article is available at: http://biorxiv.org/content/early/2016/05/13/053041.
The full citation to our paper is:
