
Together with researchers from UC San Francisco, UC Davis, Oregon State University, and the Netherlands, our group recently published a paper on bioRxiv that presents a new method capable of identifying different microbes present in human blood. Our paper is featured in the Stanford University digest of microbiome papers, as well as an international science and technology news source. Here, we demonstrate the potential use of total RNA to study the relationship of specific diseases with microbes that inhabit the human body.
A growing body of evidence suggests that the human microbiome plays an important role in health and disease. In order to investigate the specific ways that microbes may influence disease development, we developed a novel ‘lost and found’ pipeline. Here, we use whole blood RNA sequencing (RNA-Seq) reads to detect a variety of microbial organisms. Our ‘lost and found’ pipeline utilizes high quality reads that fail to map to the human genome as candidate microbial reads. Since RNA-Seq has become a widely used technology in recent years with many large datasets available, we believe that our pipeline has great potential for application across tissues and disease types.
We applied our ‘lost and found’ pipeline to study the composition of blood microbiome in almost two hundred individuals, including healthy control individuals and patients with schizophrenia, bipolar disorder, and amyotrophic lateral sclerosis (also known as ALS or Lou Gehrig’s disease). Using this pipeline, we detected bacterial and archaeal phyla in blood using RNA sequencing (RNA-Seq) data. Our analyses of these data, including examination of positive and negative control datasets, suggest that detected phyla are in fact representative of the actual microbial communities in the individuals’ blood.
In total, we observed 23 distinct microbial phyla with on average 4.1 ± 2.0 phyla per individual. Phylogenetic classification is performed using Phylosift, which assigns the filtered candidate microbial reads to the microbial genes from 23 distinct taxa on the phylum level. The large majority of taxa that were observed in our sample are not universally present in all individuals, except for Proteobacteria that dominate all samples with 73.4% ± 18.3% relative abundance (dark green color). Here, we can see the genomic abundances of microbial taxa at phylum level of classification for each of the four groups:
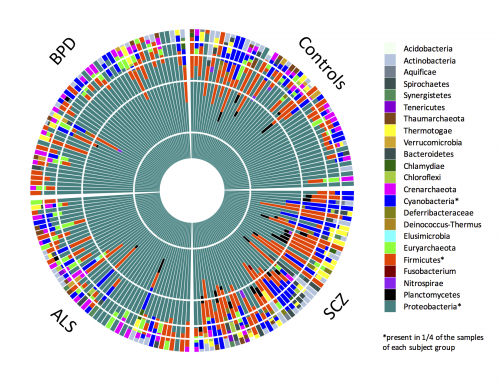
Further, in comparison to individuals in the other three groups, we observed a significantly increased microbial diversity in blood samples from individuals with schizophrenia. We replicated this finding with an independent schizophrenia case-control study. The increased microbial diversity observed in schizophrenia could be part of the disease etiology (i.e., causing schizophrenia) or may be a secondary effect of disease status. In the absence of a direct link with genetic susceptibility and the reported correlation with the immune system, we hypothesize that the observed effect in schizophrenia is secondary to disease. This phenomena may be a consequence of lifestyle differences of schizophrenia patients, including cigarette smoking, drug use, or other environmental exposures. Future targeted and/or longitudinal studies with larger sample sizes, detailed clinical phenotypes, and more in-depth sequencing are needed to corroborate this hypothesis.
We hope that our finding of increased diversity in schizophrenia will ultimately lead to a better understanding of the functional mechanisms underlying the connection between immune system, blood microbiome, and disease etiology. With the increasing availability of large scale RNA-Seq datasets collected from different phenotypes and tissue types, we anticipate that the application of our ‘lost and found’ pipeline will lead to the generation of a range of novel hypotheses, ultimately aiding our understanding of the role of the microbiome in health and disease.
This project was led by Serghei Mangul and Loes Olde Loohuis (Roel Ophoff group). This was a joint project with the Roel Ophoff group at Center for Neurobehavioral Genetics at the Semel Institute for Neuroscience and Human Behavior, University California, Los Angeles, CA, USA.
The article is available at: http://biorxiv.org/content/early/2016/06/07/057570.
The full citation to our paper is:
