The use of the multivariate normal (MVN) model has been a powerful tool in our groups research and it has been utilized in many of our papers. Jose Lozano (University of the Basque Country, San Sebastian, Spain), along with Eleazar Eskin and three ZarLab alumni—Farhad Hormozdiari (postdoc at Harvard), Jong Wha (Joanne) Joo (faculty at Dongguk University in Seoul), and Buhm Han (faculty at University of Ulsan College of Medicine in Seoul)—recently published a review of the multivariate normal (MVN) distribution framework in genome-wide association studies (GWAS) studies.
Genome-wide association studies (GWAS) have discovered thousands of variants involved in common human diseases. In these studies, frequencies of genetic variants are compared between a population of individuals with a disease (cases) and a population of healthy individual controls). Any variant that has a significantly different frequency between the two populations is considered an associated variant.
A major challenge in the analysis of GWAS studies is the fact that human population history causes nearby genetic variants in the genome to be correlated with each other. In this review, we demonstrate how to utilize the MVN distribution to explicitly take into account the correlation between genetic variants and provide a comprehensive framework for analysis of GWAS.
In this paper, we show how the MVN framework can be applied to perform association testing, correct for multiple hypothesis, testing, estimate statistical power, and perform fine mapping and imputation. In future blog posts, we will highlight different ways the MVN framework can be used in association studies.
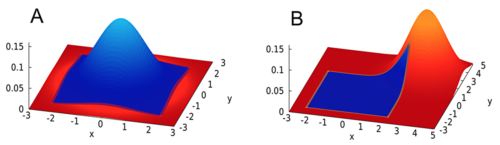
An illustration of the multivariate normal model (a) Type I Error (b) Power.
Many of the authors are the alumni of the group who pioneered the use of the MVN in various problems in association studies. Here is a list of papers that our group published using the MVN framework:
- Farhad Hormozdiari, Anthony Zhu, Gleb Kichaev, Chelsea J.-T. Ju, Ayellet V. Segre, Jong Wha J. Joo, Hyejung Won, Sriram Sankararaman, Bogdan Pasaniuc, Sagiv Shifman, and Eleazar Eskin. Widespread allelic heterogeneity in complex traits. The American Journal of Human Genetics, 100(5):789{802, may 2017.
- Yue Wu, Farhad Hormozdiari, Jong Wha J. Joo, and Eleazar Eskin. Improving imputation accuracy by inferring causal variants in genetic studies. In Lecture Notes in Computer Science, pages 303{317. Springer International Publishing, 2017.
The paper was written by Jose A. Lozano, Farhad Hormozdiari, Jong Wha (Joanne) Joo, Buhm Han, and Eleazar Eskin, and it is available at: https://www.biorxiv.org/content/early/2017/10/28/208199.
The full citation to our paper is:
Jose A. Lozano, Farhad Hormozdiari, Jong Wha (Joanne) Joo, Buhm Han, Eleazar Eskin. 2017. The Multivariate Normal Distribution Framework for Analyzing Association Studies. bioRxiv doi: https://doi.org/10.1101/208199.