
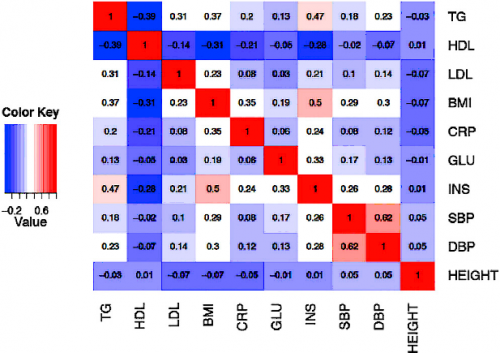
Pairwise correlation between each phenotype pair in the NFBC dataset.
In genome-wide association studies (GWAS), investigators identify variants that are significantly associated with the phenotype by collecting and performing statistical tests on genotypes and phenotypes from a set of individuals. Recently, GWAS samples have increased in size to include tens or hundreds of thousands of variants. Studies working with such large datasets have recently discovered hundreds of variants involved in multiple common diseases (Schunkert et al. 2011; Voight et al. 2010). For the most part, identified variants have very small effect sizes, suggesting that larger association studies are capable of implicating more variants.
Increasing the size of GWAS samples is a shared goal among bioinformatics researchers. Unfortunately, some phenotypes are either logistically difficult or very expensive to collect. For these phenotypes, it is impractical to perform GWAS with tens or hundreds of thousands of individuals. Examples of these difficult-to-collect phenotypes include those that require obtaining an inaccessible tissue (such as brain expression), using a complex intervention (such as a response to diet), and re-contacting individuals simply because they were unmeasured in the original cohort. For these phenotypes, an investigator finds it difficult to collect samples large enough to discover variants with small effect sizes. As a result, it is unlikely that GWAS will perform effectively on these phenotypes.
To address this issue, we developed a novel approach we call phenotype imputation. In our method, we estimate and leverage the correlation structure between multiple phenotypes to impute the uncollected phenotype. A paper presenting our approach was accepted by and is in press with the American Journal of Human Genetics.
In order to leverage the correlation structure between multiple phenotypes, we first estimate the correlation structure from a complete dataset that includes all phenotypes. We then use the conditional distribution based on the multivariate normal (MVN) statistical framework to impute the uncollected phenotypes in an incomplete dataset. Our approach uses only phenotypic—not genetic—information, enabling subsequent use of these imputed phenotypes for association testing without incurring data re-use. For GWAS including both complete and incomplete datasets, we provide an optimal meta-analysis strategy that accounts for imputation uncertainties by combining association results from both collected and imputed phenotypes. Further, our paper demonstrates that phenotype imputation can be performed using summary statistics. This result makes our method applicable to datasets where we only have access to the summary statistics and not the raw genotypes and phenotypes.
In our forthcoming AJHG paper, we use the Northern Finland Birth Cohort (NFBC) data to assess the performance of our novel method. The NFBC dataset consists of 10 phenotypes collected from 5,327 individuals. The 10 phenotypes are triglycerides (TG), highdensity lipoproteins (HDL), low-density lipoproteins (LDL), glucose (GLU), insulin (INS), body mass index (BMI), C-reactive protein (CRP) as a measure of inflammation, systolic blood pressure (SBP), diastolic blood pressure (DBP), and height. The genotype data consists of 331,476 SNPs.
Imputing the TG, BMI, and SBP phenotypes enable us to recover most of the significantly associated loci in the original data at the nominal significance level, as shown in the above figure. This result demonstrates that the imputed phenotype can effectively be used for replication purposes, even though it might not provide sufficient power for discovery purposes due to imputation uncertainties.
Our approach allows us to know the exact distribution of the imputed phenotype due to our parametric assumptions. We can directly use the mean value of this distribution as the imputed value. Furthermore, we utilize the variance of the missing phenotype in our analysis of the statistical power. The primary advantage of our framework is that it increases the power of GWASs on phenotypes that are difficult to collect. Analytical power computation is provided that allows investigators to determine the benefit of the imputation for a given dataset prospectively. Another advantage of this method is that it allows the use of summary statistics when the raw genotypes are not available.
This project was led by Farhad Hormozdiari and involved Michael Bilow. The article is available at: http://dx.doi.org/10.1016/j.ajhg.2016.04.013.
The full citation to our paper is:
